|
Hot on the trail of my post on naming, another names issue has come up. Post-molecular-biological revolution (yes, it happened), individual cells or cell types have come to be known by specific identifying (molecular) features. Neurons in the central nervous system might be described as being from a certain region, say the basal ganglia, and as being 'positive' for some receptor, for example dopamine receptor-D2. This is great even if it becomes name soup after a while, because not only does this give us a strong (and generally mutually-exclusive) functional description of a cell type, but it also outlines some of the most powerful tools we have as biologists to interrogate them.
Perhaps nowhere in biology is this naming convention more aggressive than in immunology, where even educated outside observers hear something akin to jibberish (in the technical sense), with every word seeming to start with 'CD' something or other. But if immunology has a complicated jargon, it adequately reflects the problems the field is approaching. Keeping track of the identities and functions of a limitless number of moving cells that are constantly developing in time and space and adapting to new challenges is...hard. One tool that immunologists use to identify relationships between cells is a sort of bootstrapping approach, in which it is asked which cells are 'positive' for which markers, and of those, which are positive for other markers. There are great tools to measure this in a global sense, such as flow cytometry, but assaying co-expression (i.e. co-labeling) in situ provides a special challenge. Typically in these experiments, a piece of tissue is observed, and the cells expressing markers A, B, or A+B are counted manually. This is a poor use of time, so today I set out to automate this for a grad student in the lab, with the black magic of python. Here is a starting image, taken in a real experiment, in which cells are labeled for two markers whose identity shall remain mysterious. 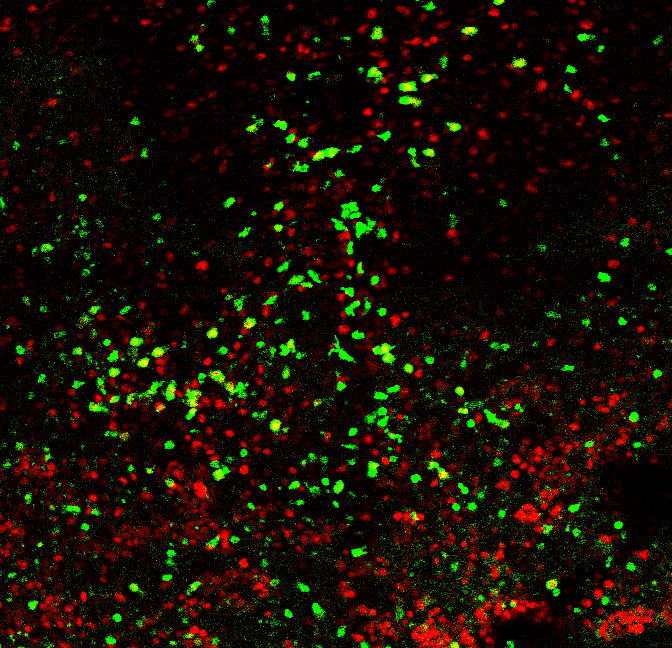
Clearly, there's a ton of background, not to mention a ton of cells. I'm going to clean this up using a median filter:
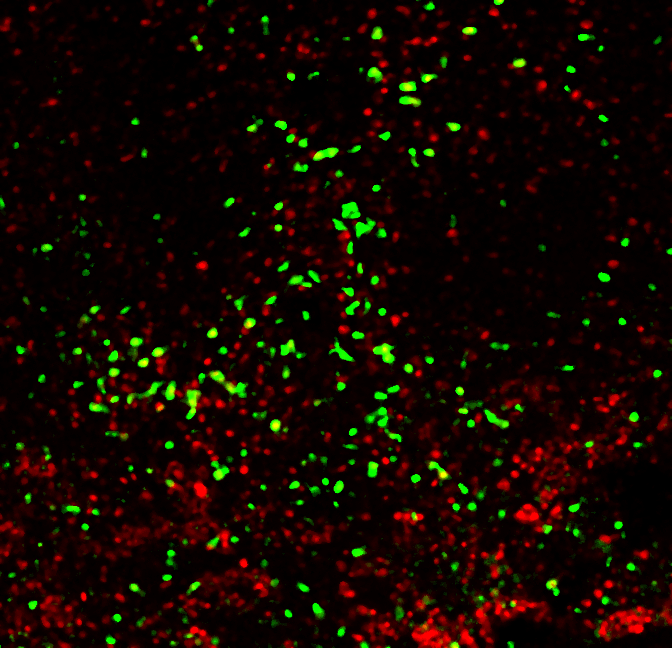
Well isn't that pretty. Now, with that black magic I mentioned above, I'm going to split the channels, find edges, filter out things that are too small or large to be single cells, then use the single-color images as a Boolean filter. Code for these operations can be found here. What comes out is a set of objects that are cell-sized and were captured in both channels. For clarity, I've overlayed it on one of the input channels.

As can be seen, the red(mask) objects overlay nicely on the native objects from this channel, providing a nice sanity check. The script also spits out summary data- how many cells per channel, and how many co-expressors.
|
 RSS Feed
RSS Feed
Proudly powered by Weebly